

点击左上角“锂电联盟会长”,即可关注!


第一作者:Matthew Li
通讯作者:Matthew Li,Khalil Amine
通讯单位:美国阿贡国家实验室
研究成果
实现锂硫(Li-S)电池的高能量密度仍然面临着巨大挑战,尤其是在痕迹电解液条件(电解液/硫质量比<3)下电解液使用量的限制,给电池性能带来了巨大的挑战,导致比容量和倍率性能非常较差。先前的研究表明,高浓度的多硫化物是造成放电电压差的原因。然而,对于在痕迹电解液条件下发生的电化学过程仍然缺乏足够的理解。
在此,美国阿贡国家实验室Khalil Amine教授和Matthew Li等人发现了一种多硫化物浓度调节机制,该机制通过沉积和再溶解固体硫基物质(可逆沉淀硫物质,RPSS)起作用。这通过正极以协调的方式发生,可以使用阻抗谱法进行测量。研究发现,形成的RPSS越多,放电的能量密度就越高。作者认为高浓度的多硫化物具有过饱和性,阻碍了RPSS的形成。使用低锂离子浓度的电解液以及使用解离一般的锂盐可以形成更多的RPSS,并最终能够在 0.05 C、>2.0 V、E/S = 2.5和室温下放电,而不使用传统的正极工程提升电池性能。
相关研究成果以“Concerted Formation of Reversibly Precipitated Sulfur Species and Its Importance for Lean Electrolyte Lithium−Sulfur Batteries”为题发表在J. Am. Chem. Soc.上。
研究背景
Li-S电池作为一种极具吸引力的下一代电池技术,它具有天然丰富的硫含量和高理论能量密度。然而,实际可实现的能量密度在很大程度上取决于所用电解液的体积/质量。在苛刻电解也条件下(电解液/硫质量比,E/S < 3),电池展现出巨大的过电位,导致极差的倍率性能(通常远低于0.05 C)。其中,提升电池性能策略围绕着从传统的Li-S电池的固-液-固转化转向固-固转化,但其最具破坏性的特性是由于反应途径的根本性变化。一项研究表明,当电池电压为<2.0 V时,电池组级能量密度会迅速下降。由于Li-S电池的工作电压徘徊在2.0~2.2 V左右,因此Li-S电池的电池组级能量密度对电池电压的任何降低/增加都特别敏感。作为商业成功的要求,保持>2.0 V 的平均放电电压,同时还要在可观的电流密度、室温、高硫含量下运行,并且所有这些都在痕迹的电解液条件下运行 (≤3.0 μL/mgs)仍然是一项艰巨的任务。
核心内容
痕迹电解液条件(LECs)的不良性能可以简单地描述为与放电过程相关的两个重要缺陷:(1)存在一个大的过电位/成核“下降”和(2)下降后的一个降低的放电电位。下降的电位通常决定了电池是否可以进入第二个放电平台(取决于截止电压)。在电池能够维持第二放电平台的情况下,被抑制的放电电位决定了电池的工作电压和能量密度。LEC性能不佳的原因可能与高多硫化物浓度有关,在第二次放电电压平台期开始之前发生的大电位骤降是预计多硫化物浓度最高的地方。传统理解认为,当多硫化物浓度增加时,粘度和相应的电解液阻抗增加,减慢了多硫化物从体相到表面的运输,或高浓度多硫化物的聚集/凝胶效应,降低反应动力学。有趣的是,虽然一些研究报告称,在最初的电位下降后,放电电位几乎完全恢复,大多数研究发现,二次放电电压处于持续低压/严重倾斜的状态。这在图1a中得到了清楚地说明,其中以传统的1 M LiTFSI DME+DOL+0.25 MLiNO3作为电解液,在第二次放电平台的整个过程中都存在明显的电压抑制。
原则上,当电池放电时,多硫化物应转化为不溶性物质(Li2S和Li2S2),多硫化物浓度应降低。如图1b所示,“有效E/S比”被估算为溶液中剩余硫质量的预期倒数,并与放电深度相对应。假定所有的硫都被消耗掉,形成不溶解的Li2S或可溶解的Li2S2。当放电深度超过第二个平台时,多硫化物浓度降低,“有效E/S比”增加。如果Li2S2是主要的放电产物,那么在电量为450 mAh g-1时,电池的多硫化物浓度预计将与注入E/S比为5 μL mg-1的“等效电池”中的最高溶解硫浓度相似。如果将Li2S视为主要产物,则电池在~700 mAh g-1时的有效E/S比为5 μL mg-1。根据文献报道,即使使用相对常规的碳纳米管/硫复合材料,E/S比为5 μL mg-1的电池预计也能在0.1 C倍率下相对无阻地运行。因此,放电时电解液中硫含量的降低应该会在一定程度上缓解电池所经历的由多硫化物浓度引起的高过电位。然而,这种情况通常不会出现,长的电压抑制会一直持续到第二个高点,而且往往根本无法恢复。因此,似乎不是高浓度的多硫化物直接导致了LEC下长的第二次平台,而是由于高浓度的多硫化物浓度的暂时存在而引起的一些负面影响。
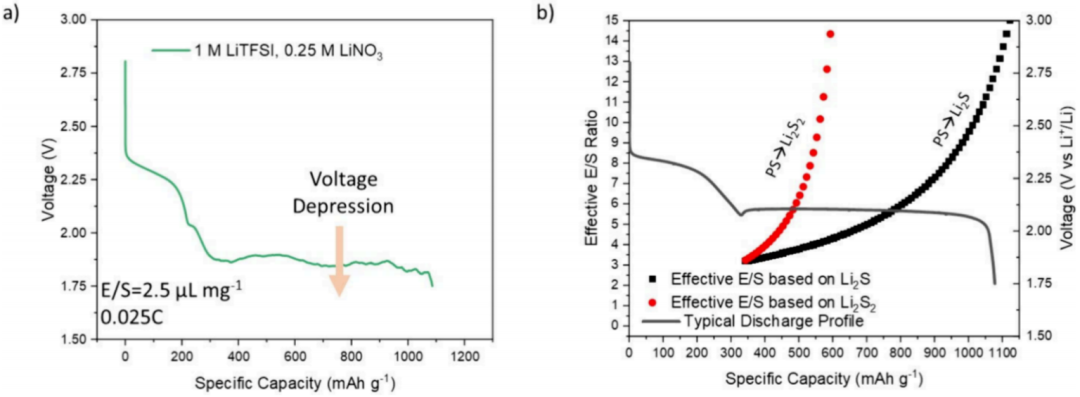
图1. 典型Li-S电池放电曲线,以及E/S比与电压的关系。
同时,放电过程中的操作电化学阻抗谱(EIS)分析可以清楚地证明过电位长期缺乏恢复。对使用不同锂盐和不同浓度的电池进行了一系列操作性EIS测量。在采用2.5 μL mgs-1的E/S比时,操作过程中的EIS(图2a)观察到了高度抑制的放电曲线。与图1a中的曲线相似,电池无法恢复循环过程中的过电位,事实上,阻抗持续增加。图2b显示了拟合阻抗值与放电时间的函数关系。Re和Rint在第二次放电平台开始时达到峰值,这与文献报道一致。进一步证实了之前的报告,在 LEC条件下,Rct是电池中最大的阻抗。与此形成鲜明对比的是,采用低锂离子成分(0.1 M LiTFSI和0.075 M LiNO3,DOL/DME 1:1 v/v)的电池产生了清晰的放电曲线,过电位显著降低(尽管仍远低于2.0 V)。
在锂离子成分低的情况下,Re峰值为~100欧姆,几乎是高浓度电解液的三倍。这种结果的原因尚不清楚,但猜测使用较低的锂离子浓度增加了溶解多硫化物的浓度,这可能导致粘度的增加。显然,低锂离子成分是可以从大过电位中恢复的电解质系统的一个例子。高浓度和低浓度的锂离子电池都能够在1.98~1.97 V的电压下进入第二放电平台。然而,较高的锂离子浓度池的电压持续下降,而低锂离子浓度的池能够保持稳定的电压。
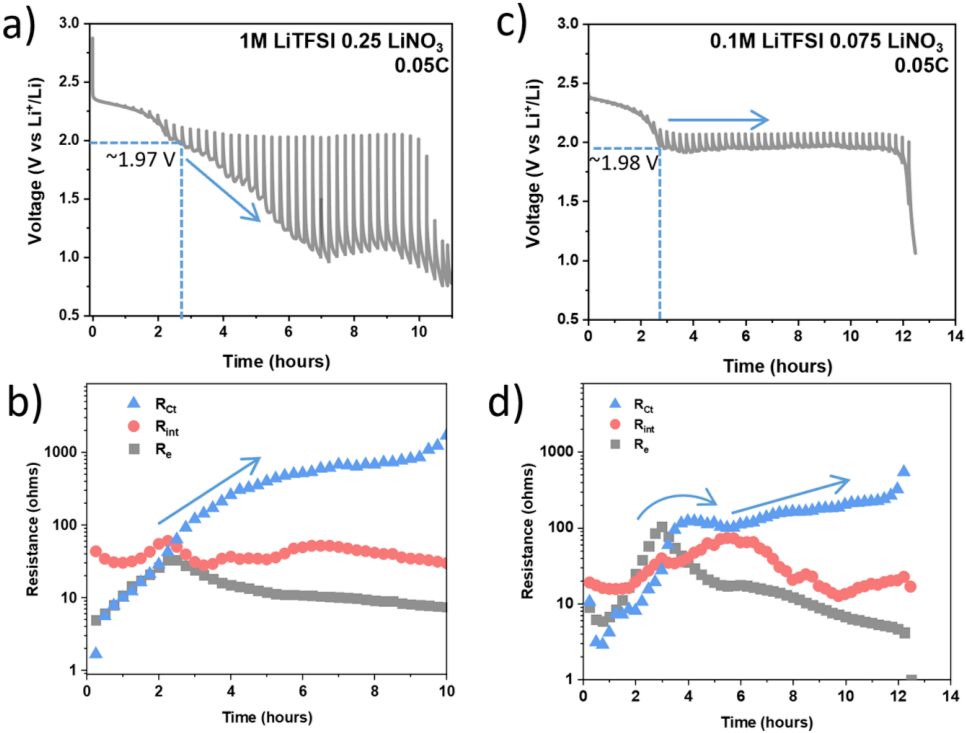
图2. 阻抗分析。
低锂离子浓度电池避免持续电压下降的能力表明,电池必须存在某种机制来管理和调节高浓度的多硫化物。为了研究这种调节机制,作者着眼于通过观察弛豫过程来了解电池在不同放电深度(DOD)下的表面状况。由于预计在LEC下的粘度和多硫化物浓度将非常高,因此猜测大部分扩散和基于溶解的弛豫过程(可用于推断正极的表面条件)发生在很长的时间尺度上,即使电池似乎必须达到平衡。为了揭示在较慢的时间尺度上发生的弛豫过程,将放电到四种不同DOD的电池在OCV下静置,同时间歇性地采样其阻抗。选择并定义了四种不同的DOD,定义如下:DOD-1:放电至8小时,图3a-c;DOD-2:放电至9小时,图3d-f;DOD-3:放电至11h,图3g-i;DOD-4:放电至13 h,图3j-l。
基于此,证实了以上的猜想,即使在OCV下静止时,阻抗的大小也发生了明显变化。由于阻抗指标趋于降低(除Rint之外),这不可能是由于锂金属腐的原因造成的。因此,从DOD-1到DOD-2再到DOD-3,弛豫趋势变化剧烈,其中DOD-4与DOD-3相对相似。这意味着电池在休息前的条件(表面、电解质成分)会随着DOD的函数而明显变化。
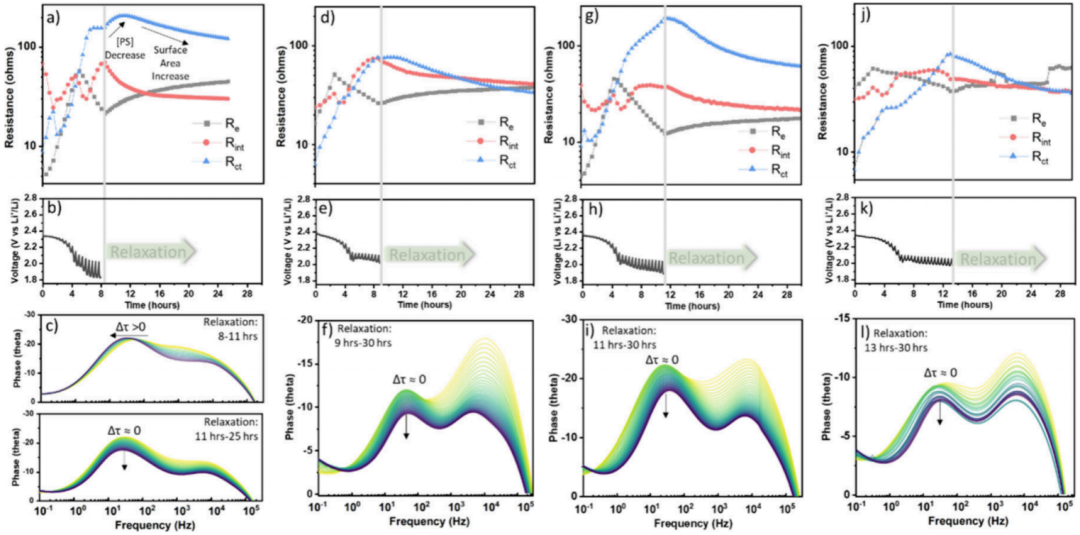
图3. EIS电池性能评估。
在DOD-1(放电8小时,图3a),与Rct相关的相位峰(~102Hz)最初转移到一个较低的频率。一旦Rct停止增加并开始减少,频率就停止移动,并开始保持~30Hz的恒定值。这表明,在最初的3h,Rct增加是由于近表面多硫化物浓度(可能扩散到电解液)减少,而随后减少Rct可以解释为暴露更多的表面积(图4c)。由于DOD-1处于第二次放电平台的早期阶段,因此可能存在非常高的多硫化物浓度。静置时,多硫化物表面浓度扩撒到电解液中,交换电流减少并 Rct增加。这表明存在一些多硫化物的固体沉淀物,当近表面多硫化物浓度降低时,这些沉淀物可以重新溶解,本文中作者将这些称为可逆沉淀硫物质(RPSS)。
放电时间较长的电池(DOD-3和DOD- 4)似乎只有Rct的下降趋势,而fct没有变化(即以表面积增加为主,图3i和3l),这同样可能是由于RPSS的溶解暴露了额外的表面,这表明当放电到DOD-3和DOD-4 时,多硫化物浓度低于表面的溶解度极限(如图 4f 所示)。当电池在DOD-2停止并休息时,Rct最初在2小时的弛豫中保持恒定值,然后继续迅速减小,类似于其他三个DOD。Rct初始常数值可能是再溶解开始的迹象,其中多硫化物浓度处于RPSS的溶解度极限,具有相当小的溶解驱动力。随着放电从DOD-1发展到DOD-2,溶液中更多的多硫化物转化为不溶性的Li2S/Li2S2。这降低了多硫化物的近表面浓度。进一步放电到DOD-3和DOD-4可能导致足够低的多硫化物浓度,使RPSS在恒电流放电停止后立即大量溶解。

图4. 该示意图说明了电池在不同DOD下的不同状态。
为了进一步了解Rint的重要性,在一系列不同电解液组成的操作EIS分析中进行了相同的分析。由于LEC对放电容量和电位都有负面影响,使用活性物质水平的能量密度(Wh kgs-1)作为主要的性能指标,与电池中的阻抗贡献相关联。正如预期的那样,第一次放电所传递的能量似乎与放电6小时时的Rct幅度呈相当强的负相关。结果显示,Rct越高,第一次放电的能量密度就越低(图5a)。值得注意的是,在第一次放电平台结束时的电解液电阻(Re)与电池的能量之间没有显著的相关性。考虑到已知的对聚硫化物浓度的依赖性,这表明由多硫化物溶解引起的电解液的高粘度可能不是一个很好的性能预测指标。
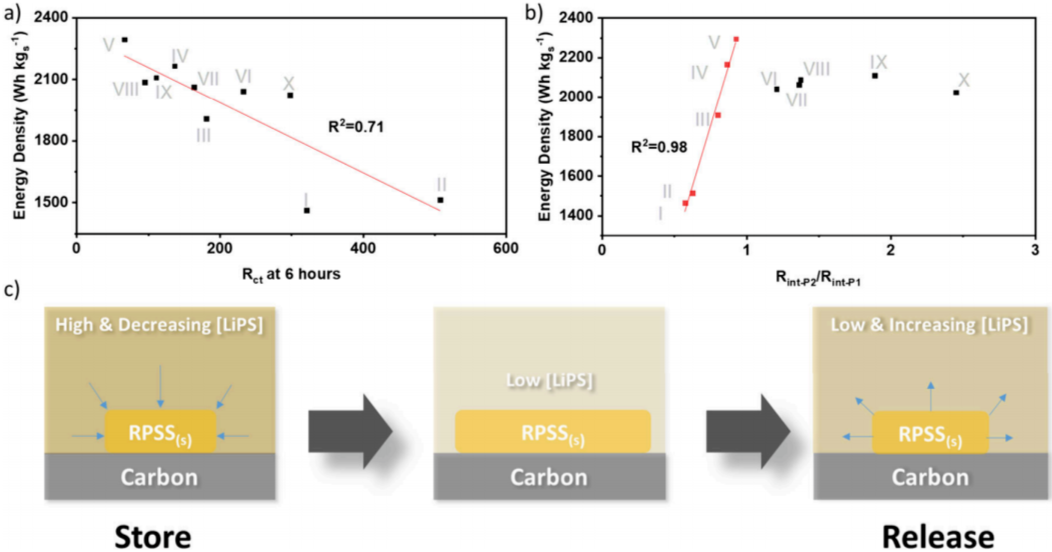
图5. 能量密度评估。
为了促进RPSS的更多形成,必须首先了解它们是如何受到限制的。众所周知,多硫化锂在高浓度或低温下往往会形成团簇。这样的集群可能无法形成RPSS,并且非常稳定。一种可能的解释是,团簇越稳定,过饱和度越高,其作为RPSS沉淀的能力就越差。对于聚合物来说尤其如此。溶液中的团簇/聚集体先前已被证明可以明显增加聚合物的溶解度。(45)相比之下,具有较不稳定的多硫化物簇的电解液往往很容易从溶液中沉淀出来,形成所需的 RPSS。为了证明这种情况,研究了聚硫化物/电解液溶液对温度的稳定性。
冷却后,预计多硫化物的溶解度会降低,这会导致沉淀。将多硫化物与高浓度和低浓度锂离子盐的电解液混合,并使用差示扫描量热法(DSC)进行研究。由于多硫化物浓度高,DOL/DME(室温下高蒸气压)蒸发的微小成分变化可能会在测试前引起过早沉淀,从而使实验结果无效。因此,溶剂改用TEGDME,其蒸气压要低得多。DSC表明,使用低锂离子浓度的电解液以及使用解离性较差的锂盐可以形成更多的RPSS,并最终在0.05C、E/S=2.5和室温下实现>2.0 V的放电,而无需使用其他的正极材料工程。
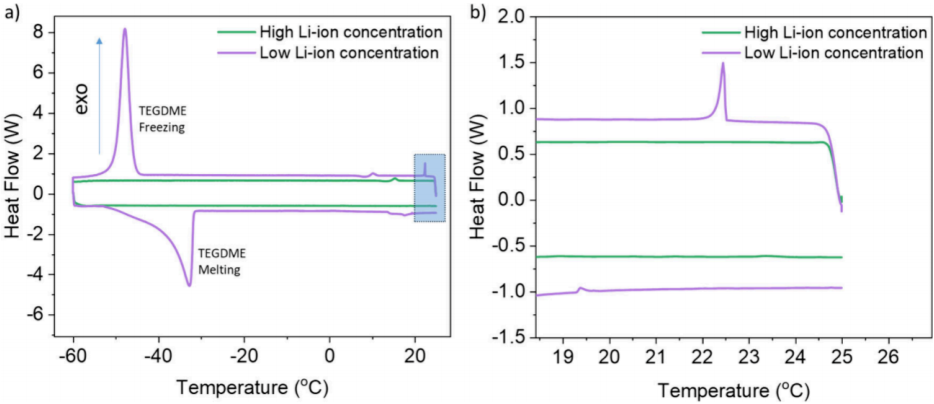
图6. 使用相同的盐和TEGDME作为溶剂,对含有高和低Li离子浓度的电解液进行差示扫描量热法。
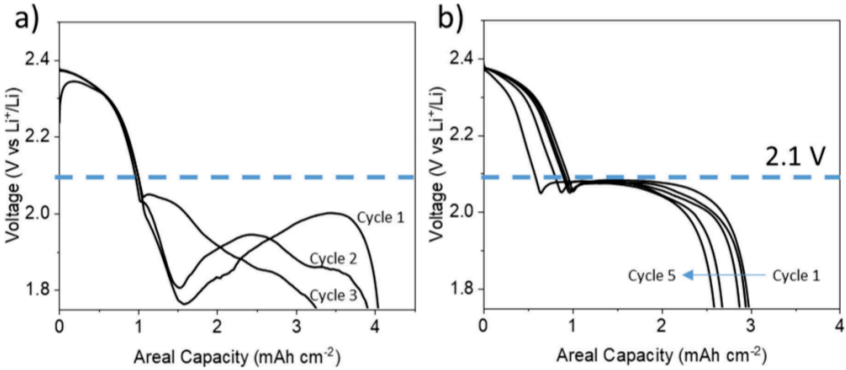
图7. 在0.05 C下循环且硫负载量为~3.5 mg cm-2的电池的放电电压曲线。
结论展望
综上所述,作者利用操作和弛豫电化学阻抗谱(EIS)分析,揭示了传统固态−液态−固态转化型Li-S电池在痕迹电解液条件下存在的多硫化物浓度调节机制,发现在特定的放电深度(DOD)下,界面阻抗(高频半圆,中心在103~105Hz)与放电材料水平的能量密度之间有很强的关系。研究发现,根据DOD的函数关系,发现具有一次以上界面阻抗增加的电池,其放电能量密度更高。作者将此归因于基于弛豫的EIS研究(其中跟踪了相角的变化)中推断出的可逆沉淀的硫基物种。同时,坐着讨论了可逆沉淀硫(RPSS)的形成及其重要性,促进RPSS的形成可显著提高电池的电位和容量。使用锂离子高度缺乏且阴离子解离能力差的电解液和标准的“非工程”硫电极,即市场上可买到的微米级硫颗粒(3. 5 mg cm-2)(硫含量为 60%)的标准“非工程”硫电极,能够在室温和E/S比为2.5下以0.05 C(1 C = 1675 mA g-1)的倍率实现高放电比容量(~850 mAh g-1),第二平台远高于2.0 V。
文献信息
Matthew Li,* Xiaozhou Huang, Chi Cheung Su, and Khalil Amine*, Concerted Formation of Reversibly Precipitated Sulfur Species and Its Importance for Lean Electrolyte Lithium−Sulfur Batteries, J. Am. Chem. Soc.
https://doi.org/10.1021/jacs.4c05000
